Vector Reorientation Dynamics (VRD)#
This notebook demonstrates how to compute the reorientation autocorrelation function Cˡ(t) of a molecular vector (e.g. an O–H bond or a C–C bond) and extract its characteristic reorientation time τ.
Theory — Legendre polynomial autocorrelation:
where \(\hat{\mathbf{u}}(t)\) is the unit vector at time t and \(P_l\) is the l-th Legendre polynomial:
l |
\(P_l(x)\) |
Experiment |
|---|---|---|
1 |
\(x\) |
— |
2 |
\(\tfrac{1}{2}(3x^2-1)\) |
NMR T₁, dielectric relaxation |
3 |
\(\tfrac{1}{2}(5x^3-3x)\) |
Raman / IR |
Cˡ decays from 1 to 0. At long times it is fitted with the Kohlrausch–Williams–Watts (KWW) stretched exponential:
Topics covered:
O–H bond reorientation dynamics of water molecules (l = 1)
C–C bond reorientation of trifluoroacetate (TFA) anions (l = 3)
Comparing individual-bond vs. ensemble-averaged Cˡ(t)
KWW fitting to extract τ and β
import numpy as np
import matplotlib.pyplot as plt
from fishmol import trj, funcs, style
from cage_data import cage1_info
Read trajectory file#
%%time
cell = cage1_info.cell
# cell is a fallback: this NVT file has no Lattice= in frame comments
traj = trj.Trajectory(timestep = 5, data = "/nobackup/rhtp48/data_ana/cage1-500K.xyz", index = ":", cell = cell)
CPU times: user 2min 17s, sys: 7.39 s, total: 2min 24s
Wall time: 2min 27s
O–H Bond Reorientation of Water Molecules#
funcs.VRD parameters:
traj—Trajectoryobjectspec— atom specification:[donor_idx, [H1_idx, H2_idx, …]]for multiple vectors from one atom, or[[g1_indices], [g2_indices]]for paired groupsnum— number of frames per chunk (window length for time-origin averaging)sampling— stride within each chunk (every n-th frame is used)skip— stride across chunks (every n-th chunk is used for averaging)l— Legendre polynomial order (1, 2, or 3)mean— ifTrue, average Cˡ over all vector pairs; ifFalse, return one curve per pairfit— ifTrue, fit the decay with KWW and plot the result
Sepearately calculate the O-H bonds#
water_vrd = funcs.VRD(traj = traj, spec = [14, [15, 16]], num = 500, sampling = 5, skip = 2)
results = water_vrd.calculate(l = 1, mean = False, fit = True, plot = True)
Progress: [■■■■■■■■■■■■■■■■■■■■] 100.0%
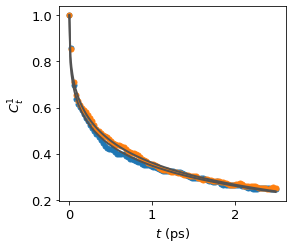
Average the O-H bonds of the same water molecule#
water_vrd = funcs.VRD(traj = traj, spec = [14, [15, 16]], num = 500, sampling = 5, skip = 2)
results = water_vrd.calculate(l = 1, mean = True, fit = True, plot = True)
Progress: [■■■■■■■■■■■■■■■■■■■■] 100.0%
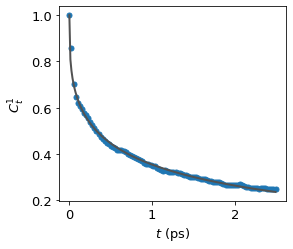
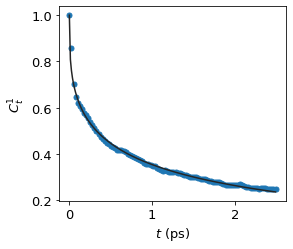
Plot the results by yourself#
fig, ax = plt.subplots()
ax.scatter(results.t, results.C_t)
ax.plot(results.t_fit, results.C_t_fit, color = "#252525")
ax.set_xlabel(r"$t$ (ps)")
ax.set_ylabel(r"$C^1_t$")
plt.show()
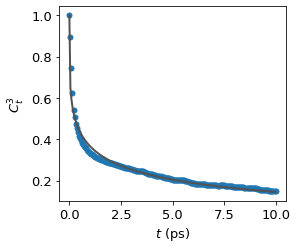
TFA O···O Vector Reorientation (l = 3)#
For TFA anions we track the vector connecting the two carboxylate oxygens (O1···O2), which is a proxy for the tumbling of the CF₃COO⁻ ion. Because this vector is along the C–C bond axis, its reorientation reflects the overall rotational motion of the anion.
We use l = 3, which probes the same dynamics as Raman spectroscopy and is more sensitive to fast librational motion than l = 2.
Single TFA — individual O···O vector#
Calculate the C-C bond reorientation dynamics in one TFA anion#
TFAs = cage1_info.TFAs
spec = [*TFAs[0].values()]
spec
[0, 1, 2, 3, 4, 5, 6]
print([x for x in spec if traj.frames[0][x].symbs == "O"])
[3, 4]
vrd = funcs.VRD(traj=traj, spec=[3, 4], num=2000, sampling=10, skip=5)
results = vrd.calculate(l=3, mean=False, fit=True, plot=True)
all_oxygen = [x for spec in TFAs for x in [*spec.values()] if traj.frames[0][x].symbs == "O"]
specs = [[x for x in all_oxygen[::2]], [x for x in all_oxygen[1::2]]]
specs
[[3, 10, 132, 139, 261, 268, 390, 397], [4, 11, 133, 140, 262, 269, 391, 398]]
All TFA ions — ensemble average#
By passing two lists of atom indices (one per end of the vector) funcs.VRD computes Cˡ for every O1ᵢ···O2ᵢ pair in parallel and averages them. This reduces statistical noise and gives the ensemble-average reorientation time of the TFA anion.
vrd = funcs.VRD(traj = traj, spec = specs, num = 2000, sampling = 10, skip = 5)
results = vrd.calculate(l = 3, mean = True, fit = True, plot = True)
Progress: [■■■■■■■■■■■■■■■■■■■■] 100.0%